
Hemofilia
Sabías ...
SOS Children, una organización benéfica educación , organizó esta selección. Madres SOS cada aspecto después de un una familia de niños apadrinados .
| Hemofilia d | |
|---|---|
| Clasificación y recursos externos | |
| CIE- 10 | D 66- D 68 |
| CIE- 9 | 286 |
| OMIM | 306700 306900 264900 |
| DiseasesDB | 5555 5561 29376 |
| MedlinePlus | 000537 |
| Medcenter | med / 3528 |
| MeSH | D025861 |
Hemofilia (también deletreado como la hemofilia, del griego haima "sangre" y philia "amar") es un grupo de hereditario trastornos genéticos que alteran la capacidad del cuerpo para controlar la sangre de coagulación o coagulación. En su forma más común, La hemofilia A, la coagulación factor VIII está ausente. En La hemofilia B, factor IX es deficiente. La hemofilia A se presenta en aproximadamente 1 de cada 5.000-10.000 nacimientos de varones, mientras que la hemofilia B ocurre en aproximadamente 1 en aproximadamente 20,000-34,000.
Los efectos de esta ligada al sexo, Trastorno cromosoma X se manifiestan casi en su totalidad en los hombres, aunque el gen para el trastorno se hereda de la madre. Las mujeres tienen dos cromosomas X, mientras que los hombres tienen sólo uno, que carece de una "copia de seguridad" copia del gen defectuoso. Las hembras son, por tanto, casi exclusivamente los portadores de la enfermedad, y pueden haber heredado de cualquiera de su madre o padre. En aproximadamente el 30% de los casos de hemofilia B, sin embargo, no hay antecedentes familiares de la enfermedad y la condición es el resultado de una mutación genética espontánea. Una madre que es una portadora tiene un 50% de heredar el cromosoma X defectuoso a su hija, mientras que un padre afectado siempre transmitir el gen afectado a sus hijas. Un hijo no puede heredar el gen defectuoso de su padre.
Estas deficiencias genéticas pueden reducir el plasma sanguíneo los niveles de factor de coagulación de los factores de coagulación necesarios para un proceso de coagulación normal. Cuando un vaso sanguíneo se lesiona, una costra temporal hace formar, pero los factores de coagulación que faltan a prevenir la formación de fibrina que es necesario para mantener el coágulo de sangre. Así, un hemofílico no sangra más intensamente que una persona normal, pero por una cantidad mucho mayor de tiempo. En hemofílicos graves, incluso una lesión menor puede resultar en la pérdida de sangre que dura días, semanas, o no siempre la curación por completo. El riesgo fundamental aquí es normalmente con pequeñas lesiones que, debido a la falta de factor VIII, obtener tiempos largos para sanar. En áreas tales como el cerebro o en el interior articulaciones esto puede ser mortal o debilitante forma permanente.
El sangrado con externa lesión es normal, pero la incidencia de la tarde re-sangrado y hemorragia interna se incrementa, especialmente en los músculos, articulaciones, o sangrado en espacios cerrados. Las complicaciones mayores incluyen hemartrosis, hemorragia , sangrado gastrointestinal, y menorragia.
Causas
La hemofilia es casi siempre causada por un error genético que causa la falta de un factor de coagulación que funciona normalmente:
- La hemofilia A implica una falta de coagulación funcional Factor VIII. (Esto representa 90% de los casos de hemofilia.)
- La hemofilia B implica una falta de coagulación funcional Factor IX.
- La hemofilia C implica una falta de coagulación funcional Factor XI.
- Hipofibrinogenemia implica una falta de factor de coagulación funcional I. Debido a que es tan rara, alrededor de 1 a 2 casos por cada millón de nacimientos, no tiene tratamiento definitivo aprobado por la FDA. Afecta a hombres y mujeres por igual. La sangre de las personas con Hipofibrinogenemia ni coágulos, ni contiene cantidades suficientes de El fibrinógeno.
Aparición
La hemofilia es bastante raro, con sólo alrededor de 1 instancia de cada 10.000 nacimientos (o 1 de cada 5.000 nacimientos de varones) para la hemofilia A y 1 de cada 50.000 nacimientos para la hemofilia B. Cerca de 18,000 personas en los Estados Unidos tienen hemofilia. Cada año en los EE.UU., a unos 400 bebés nacen con este trastorno. La hemofilia ocurre generalmente en los hombres y con menos frecuencia en las mujeres. Se estima que alrededor de 2500 los canadienses tienen hemofilia A y alrededor de 500 canadienses tienen hemofilia B.
Historia
La referencia implícita más temprana posible para la hemofilia puede haber estado en la Talmud, un judío texto sagrado, que dice que los hombres no tenían que ser circuncidados si dos hermanos ya habían muerto a causa de la intervención. En 1000, la Médico árabe Abulcasis (conocido como Albucasis en Occidente) escribió una descripción más explícita de la hemofilia en su Al-Tasrif, en la que escribió de un Familia andaluza cuyos machos murió de hemorragia después de lesiones menores.
En 1803 , el Dr. John Conrad Otto, un médico de Filadelfia, escribió un relato sobre "una disposición hemorrágica existente en ciertas familias." Reconoció que el trastorno era hereditario y que afectó a varones y hembras raramente. Él fue capaz de rastrear la enfermedad de nuevo a una mujer que se estableció cerca de Plymouth en 1720. El primer uso del término "hemofilia" aparece en una descripción de la condición escrita por Hopff en la Universidad de Zurich en 1828 . En 1937 , Patek y Taylor, dos médicos de Harvard, descubrió globulina anti-hemofílico. Pavlosky, un médico de Buenos Aires, que se encuentra La hemofilia A y La hemofilia B para ser enfermedades separadas por hacer una prueba de laboratorio. Esta prueba se realiza mediante la transferencia de la sangre de un hemofílico a otro hemofílico. El hecho de que esto solucione el problema de coagulación mostró que había más de una forma de la hemofilia.
Hemofilia en la realeza europea un lugar destacado y por lo tanto a veces se conoce como "la enfermedad real". Reina Victoria pasó la mutación a su hijo Leopold y, a través de varias de sus hijas, a varios miembros de la realeza de todo el continente, incluyendo las familias reales de España, Alemania, y Rusia. Zarevich Alexei Nikolaevich, hijo de Nicolás II, era descendiente de la reina Victoria y sufría de hemofilia. Se alegó que Rasputin fue un éxito en el tratamiento de la Zarevich Alexei de hemofilia de Rusia. En ese momento, un tratamiento común administrado por médicos profesionales fue usar aspirina, que empeora en lugar de disminuir el problema. Se cree que, simplemente desaconsejando el tratamiento médico, Rasputín podría traer una mejora visible y significativa a la condición de Alexei.
Antes de 1985, no había leyes promulgadas dentro de los EE.UU. para detectar sangre. Como resultado, muchos pacientes con hemofilia que recibieron factor de coagulación no probado y no apantallado antes de 1992 estaban en un riesgo extremo de contraer el VIH y Hepatitis C a través de estos productos de la sangre. Se estima que más del 50% de la población de la hemofilia, más de 10.000 personas, contrajo el VIH desde el suministro de sangre contaminada en los Estados Unidos solamente.
Como resultado directo de la contaminación del suministro de sangre a finales de 1970 y principios de mediados de 1980 / con virus como el Hepatitis y VIH , los nuevos métodos se desarrollaron en la producción de productos de factor de coagulación. La respuesta inicial fue tratar térmicamente ( pasteurizar) concentrado de factor derivado de plasma, seguido por el desarrollo del factor monoclonal concentrados, que utilizan una combinación de tratamiento térmico y cromatografía de afinidad para inactivar cualquier agentes virales en el plasma reunido a partir del cual se deriva el concentrado de factor. La Lindsay Tribunal en Irlanda investigó, entre otras cosas, la lenta adopción de los nuevos métodos.
Genética
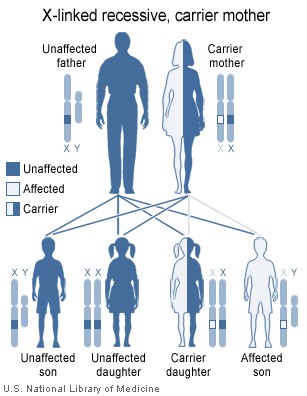
Las hembras poseen dos cromosomas X, mientras que los hombres tienen un X y un Cromosoma Y. Dado que las mutaciones que causan la enfermedad son recesiva, una mujer que lleva el defecto en uno de sus cromosomas X no pueden verse afectados por el mismo, como el equivalente alelo en su otro cromosoma debe expresarse para producir los factores de coagulación necesarios. Sin embargo, el cromosoma Y en los hombres no tiene gen de los factores VIII o IX. Si los genes responsables de la producción de factor VIII o factor IX presente en el cromosoma X de un varón son deficientes no hay equivalente en el cromosoma Y, por lo que el gen deficiente no está enmascarado por la alelo dominante y se desarrollará la enfermedad.
Desde un varón recibe su único cromosoma X de su madre, el hijo de una mujer sana en silencio que lleva el gen deficiente tendrá un 50% de probabilidades de heredar dicho gen de ella y con ella la enfermedad; y si su madre se ve afectada con hemofilia, tendrá una probabilidad del 100% de ser hemofílico. Por el contrario, para una mujer herede la enfermedad, ella debe recibir dos cromosomas X deficientes, uno de su madre y el otro de su padre (por lo tanto, que debe ser un hemofílico sí mismo). De ahí que la hemofilia es mucho más común entre los hombres que entre las mujeres. Sin embargo, es posible que las mujeres portadoras para convertirse en hemofílicos leves debido a lyonisation (inactivación) de los cromosomas X. Hijas hemofílicos son más comunes de lo que eran, como mejores tratamientos para la enfermedad han permitido más hemofílico varones de sobrevivir hasta la edad adulta y convertirse en padres. Las hembras adultas pueden experimentar menorragia (menstruación abundante), debido a la tendencia a la hemorragia. El patrón de herencia es de tipo cruzado. Este tipo de patrón también se observa en la ceguera al color .
Como con todos los trastornos genéticos, es, por supuesto, también es posible para un ser humano de adquirir espontáneamente a través mutación, en vez de heredarla, debido a una nueva mutación en uno de los gametos de sus padres. Las mutaciones espontáneas representan aproximadamente el 33% de toda la hemofilia A y el 20% de toda la casos de hemofilia B. Las pruebas genéticas y Se recomienda la asesoría genética para las familias con hemofilia. Las pruebas prenatales, como amniocentesis, está disponible para las mujeres embarazadas que pueden ser portadores de la enfermedad.
Probabilidad
Si una mujer da a luz a un niño hemofílico, ya sea la hembra es portadora de la enfermedad o la hemofilia fue el resultado de un mutación espontánea. Hasta moderna directo Las pruebas de ADN, sin embargo, fue imposible determinar si una mujer con sólo los niños sanos fue de un portador o no. En general, los hijos más sanos parió, mayor será la probabilidad de que ella no era portadora. Si el Factor RH del varón nacido es diferente de la madre, el niño no se verá afectada.
Si un hombre está afectado por la enfermedad y tiene hijos, sus hijas serán portadoras de hemofilia. Sus hijos, sin embargo, no se verán afectados con la enfermedad. Esto se debe a que la enfermedad está ligada al X y el padre no puede pasar a través de la hemofilia cromosoma Y. Los hombres con este trastorno son entonces no más probabilidades de transmitir el gen a sus hijos que las mujeres portadoras, aunque todos hija que engendran será transportistas y todos los hijos que el padre no tenga hemofilia (a menos que la madre es portadora).
Tratamiento
Aunque no existe una cura para la hemofilia, que se puede controlar con infusiones regulares del factor de coagulación deficiente, es decir, factor VIII en la hemofilia A o factor IX en la hemofilia B. Factor de reemplazo puede ser aislado de humano suero de la sangre, recombinante, o una combinación de los dos. Algunos hemofílicos desarrollar anticuerpos (inhibidores) contra los factores de sustitución que se les da, por lo que la cantidad del factor tiene que ser aumentado o productos de reemplazo no humanos deben ser determinada, como factor VIII porcino.
Si un paciente llega a ser refractarios a factor de coagulación de reemplazo como resultado de inhibidores circulantes, esto puede superarse parcialmente con humana recombinante factor VII (NovoSeven), registrada para esta indicación en muchos países.
A principios de 2008, los EE.UU. Food and Drug Administration aprobó Xyntha ( Wyeth) factor anti-hemofílico, ingeniería genética a partir de los genes de las células de ovario de hámster chino. Desde 1993 (la doctora Mary Nugent) productos de factores recombinantes (que suelen ser cultivadas en células de ovario de hámster chino ( CHO) y células de cultivo de tejidos implican poco, si alguno productos de plasma humano) han estado disponibles y han sido ampliamente utilizados en los países occidentales más ricos. Mientras que los productos de factor de coagulación recombinante ofrecen una mayor pureza y seguridad, que son, como concentrado, muy caro, y por lo general no están disponibles en el mundo en desarrollo. En muchos casos, los productos de factor de cualquier tipo son difíciles de obtener en los países en desarrollo.
En los países occidentales, las normas comunes de atención caen en una de dos categorías: profilaxis o bajo demanda. La profilaxis consiste en la infusión de factor de coagulación en un horario regular para mantener los niveles de coagulación suficientemente alta para evitar episodios hemorrágicos espontáneos. En la demanda de tratamiento implica el tratamiento de los episodios de sangrado, una vez que se presenten. En 2007, un estudio clínico se publicó en el New England Journal of Medicine (NEJM) comparar el tratamiento a demanda de los niños (<30 meses) con hemofilia A con tratamiento profiláctico (infusiones de 25 UI / kg de peso corporal Factor VIII cada dos días) con respecto a su efecto en la prevención de enfermedades de las articulaciones. Cuando los chicos llegaron a los 6 años de edad, el 93% de los del grupo de profilaxis y el 55% de aquellos en el grupo de terapia episódica tenían una estructura conjunta índice normal MRI. El tratamiento profiláctico, sin embargo, resultó en promedio costos de $ 300.000 por año. El autor de un editorial publicado en la misma edición de la NEJM exige más estudios clínicos que abordan la relación coste-efectividad del tratamiento profiláctico.
Se recomienda que las personas afectadas por hemofilia hacen ejercicios específicos para fortalecer las articulaciones, como los codos, las rodillas y los tobillos. Los ejercicios incluyen elementos que aumentan la flexibilidad, el tono y la fuerza de los músculos, aumentando su capacidad de proteger las articulaciones de hemorragias dañinas. Se recomiendan estos ejercicios después de que ocurre una hemorragia interna y en una base diaria para fortalecer los músculos y las articulaciones para evitar nuevos problemas de sangrado. Muchos ejercicios recomendados incluyen deportes estándar de calentamiento y ejercicios de entrenamiento como el estiramiento de las pantorrillas, los círculos de tobillo, las flexiones de codo, y conjuntos de cuádriceps.
Los tratamientos alternativos y complementarios
Los estudios científicos indican que la hipnosis y la autohipnosis pueden ser eficaces para reducir las hemorragias y la gravedad de las hemorragias y por lo tanto la frecuencia del tratamiento de los factores. Hierbas que fortalecen los vasos sanguíneos y actúan como astringentes también pueden beneficiar a los pacientes con hemofilia. Estas hierbas incluyen: Arándano ( Vaccinium myrtillus), extracto de semilla de uva ( Vitis vinifera), retama negra ( Cytisus scoparius), ortiga ( Urtica dioica), hamamelis ( Hamamelis virginiana), y milenrama ( Achillea millefolium).
Diagnóstico diferencial
La hemofilia A puede ser imitado por Enfermedad de von Willebrand
- tipo de enfermedad de von Willebrand 2A, donde disminución de los niveles de von Willebrand Factor puede conducir a la proteolisis prematura de Factor VIII. A diferencia de la hemofilia, enfermedad de von Willebrand tipo 2A se hereda en un manera autosómica dominante.
- Tipo de enfermedad de von Willebrand 2N, donde von Willebrand Factor no puede obligar Factor VIII, herencia autosómica recesiva. (Es decir, ambos padres tienen que dar al niño una copia del gen).
- tipo de enfermedad de von Willebrand 3, donde la falta de von Willebrand Factor causa proteólisis prematura de Factor VIII. A diferencia de la hemofilia, enfermedad de von Willebrand tipo 3 se hereda de forma autosómica recesiva.