
La drépanocytose
Saviez-vous ...
SOS Enfants, qui se déroule près de 200 sos écoles dans le monde en développement, a organisé cette sélection. Pour comparer les organismes de bienfaisance de parrainage ce est le meilleur lien de parrainage .
| La drépanocytose | |
|---|---|
| les ressources de classification et externes | |
 La figure (A) montre des globules rouges normaux se écoulant librement à travers les veines. L'encart montre une coupe transversale d'un globule rouge à l'hémoglobine normale normale. Figure B montre des cellules anormales, les globules rouges falciformes log brouillage, coller et accumuler au point dans une veine de branchement. L'image en médaillon montre une coupe transversale d'une drépanocytose avec de longs brins HbS polymérisés étirement et déformer la forme de la cellule. | |
| CIM 10 | Ré 57 |
| CIM 9 | 282,6 |
| OMIM | 603903 |
| DiseasesDB | 12069 |
| MedlinePlus | 000527 |
| eMedicine | med / 2126 oph / 490 ped / 2096 Emerg / 26 Emerg / 406 |
| MeSH | C15.378.071.141.150.150 |
| GeneReviews |
|
La drépanocytose (SCD), ou la drépanocytose (SCA) ou la drépanocytose, est une autosomique génétique récessif trouble sanguin avec surdominance, caractérisé par les globules rouges qui supposent une anormale, rigide, forme de faucille. Sickling diminue la flexibilité et les résultats des cellules dans un risque de complications diverses. La falciformation produit en raison d'un mutation dans le hémoglobine gène. L'espérance de vie est raccourcie. En 1994, aux États-Unis, l'espérance de vie moyenne des personnes atteintes de cette condition a été estimée à 42 années chez les hommes et 48 ans chez les femelles, mais aujourd'hui, grâce à une meilleure gestion de la maladie, les patients peuvent vivre dans leurs années 50 ou au-delà.
La drépanocytose est plus fréquente chez les personnes (ou leurs descendants) à partir de pièces de tropicale et sub-tropicale régions subsaharienne où le paludisme est, ou était commun. Dans les zones où le paludisme est endémique, il ya un prestations de remise en forme dans l'exercice qu'un gène de la drépanocytose unique ( trait drépanocytaire). Ceux avec un seul des deux allèles de la drépanocytose, sans totalement résistantes, sont plus tolérants à l'infection et ainsi montrer des symptômes moins sévères lorsqu'ils sont infectés.
La drépanocytose est le nom d'une forme spécifique de la drépanocytose dans lequel il est homozygotie pour la mutation qui provoque HbS. La drépanocytose est aussi appelé "HbSS», «maladie SS", "l'hémoglobine S" ou permutations de celui-ci. En personnes hétérozygotes, qui ont un seul gène de faucille et un gène de l'hémoglobine adulte normale, il est appelé "HbAS" ou "trait drépanocytaire". D'autres formes plus rares de la drépanocytose comprennent faucille maladie de l'hémoglobine C (HBSC), faucille bêta-Plus- thalassémie (HbS / β +) et la faucille bêta-thalassémie zéro (HbS / β 0). Ces autres formes de maladie à hématies falciformes sont États hétérozygotes composés dans lesquels la personne a une seule copie de la mutation qui provoque HbS et une copie d'un autre anormale hémoglobine allèle.
La maladie de terme est appliqué, parce que le anomalie héréditaire provoque un état pathologique qui peut conduire à la mort et de graves complications. Pas toutes les variantes héritées de hémoglobine sont préjudiciables, un concept connu sous le nom polymorphisme génétique.
Signes et symptômes
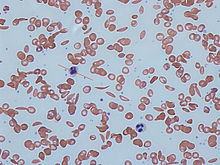
La drépanocytose peut conduire à diverses complications aiguës et chroniques, dont plusieurs ont un taux de mortalité élevé.
Crise de drépanocytose
Le terme «crise de drépanocytose» est utilisé pour décrire plusieurs conditions aiguës indépendants survenant chez les patients atteints de la drépanocytose. Sickle résultats de la maladie de la cellule à l'anémie et les crises qui pourraient être de plusieurs types, y compris le crise vaso-occlusive, crise aplasique, crise de la séquestration, crise hémolytique et d'autres. La plupart des épisodes de crises drépanocytaires durent entre cinq et sept jours. "Bien que l'infection, la déshydratation et l'acidose (tous qui favorisent falciformation) peut agir comme déclencheurs, dans la plupart des cas, aucune cause prédisposante est identifié."
Crise vaso-occlusive
Le crise vaso-occlusif est provoquée par les globules rouges en forme de faucille qui obstruent les capillaires et restreindre le flux sanguin à un organe, d'où l'ischémie, la douleur, nécrose et des dommages souvent d'organes. La fréquence, la gravité et la durée de ces crises varient considérablement. Crises douloureuses sont traités avec hydratation, analgésiques, et la transfusion sanguine; gestion de la douleur nécessite l'administration d'opiacés à intervalles réguliers jusqu'à ce que la crise se est installée. Pour crises plus doux, un sous-groupe de patients à gérer sur AINS (tels que diclofénac ou naproxen). Pour des crises plus graves, la plupart des patients ont besoin de prise en charge hospitalière des opiacés par voie intraveineuse; analgésie dispositifs (PCA) de contrôlée par le patient sont couramment utilisés dans ce cadre. Crise vaso-occlusive impliquant des organes tels que le pénis ou les poumons sont considérés comme une urgence et traité avec des transfusions de globules rouges de sang. Diphenhydramine est parfois efficace pour les démangeaisons associées à l'utilisation d'opioïdes. Spirométrie incitative, une technique visant à encourager la respiration profonde pour minimiser le développement de atélectasie, est recommandée.
Splénique crise de séquestration
En raison de ses vaisseaux étroits et la fonction de nettoyage des globules rouges sanguins défectueux, les la rate est souvent affectée. Il est habituellement infarci avant la fin de l'enfance chez les personnes souffrant de la drépanocytose. Cette autosplenectomy augmente le risque d'infection par organismes encapsulés; antibiotiques préventifs et les vaccins sont recommandés pour ceux avec une telle asplénie.
- Spléniques crises de séquestration: sont, élargissements aigus et douloureux de la rate, causées par le piégeage intrasplénique de globules rouges et entraînant une chute brutale des taux d'hémoglobine avec le potentiel de choc hypovolémique. crises de séquestration sont considérés comme une urgence. Si ne est pas traitée, les patients peuvent mourir dans les 1-2 heures en raison d'une insuffisance circulatoire. La direction est favorable, parfois avec la transfusion sanguine. Ces crises sont transitoires, ils continuent pendant 3-4 heures et peuvent durer un jour.
Crise aplasique
Crises aplasique sont aggravations aiguës de l'anémie initiales du patient, produisant pâleur, tachycardie, et la fatigue. Cette crise est déclenchée normalement par parvovirus B19, qui affecte directement l'érythropoïèse (production de globules rouges) en envahissant les précurseurs des globules rouges et en multipliant en eux et de les détruire. Infection par le parvovirus presque complètement empêche production de globules rouges dans le sang pendant deux à trois jours. Chez les individus normaux, ce est de peu de conséquence, mais la vie des globules rouges raccourcie de patients drépanocytaires en résulte une situation brutale, la vie en danger. numération des réticulocytes chuter de façon spectaculaire au cours de la maladie (causant réticulocytopénie), et la rotation rapide des globules rouges conduit à la chute du taux d'hémoglobine. Cette crise prend quatre jours à une semaine à disparaître. La plupart des patients peuvent être gérés avec soutien; certains ont besoin de transfusion sanguine.
Crise hémolytique
Crises hémolytiques sont accélérées gouttes aiguës dans le niveau d'hémoglobine. Les globules rouges se décomposent à une vitesse plus rapide. Ceci est particulièrement fréquente chez les patients co-existant déficit en G6PD. La direction est favorable, parfois avec des transfusions sanguines.
Autre
Une des premières manifestations cliniques est dactylite, présentant dès six mois, et peut se produire chez les enfants atteints trait faucille. La crise peut durer jusqu'à un mois. Un autre type reconnu de crise faucille est le syndrome thoracique aigu, une affection caractérisée par de la fièvre, des douleurs thoraciques, une difficulté respiratoire et pulmonaire infiltrer sur un radiographie pulmonaire. Étant donné que la pneumonie et la falciformation dans les poumons peuvent à la fois produire ces symptômes, le patient est traité pour les deux conditions. Il peut être déclenché par la crise douloureuse, infection des voies respiratoires, l'embolisation de moelle osseuse, ou éventuellement par une atélectasie, l'administration d'opiacés, ou la chirurgie.
Complications
La drépanocytose peut conduire à diverses complications, notamment:
- Post écrasante (auto) de l'infection splénectomie (OPSI), qui est due à une asplénie fonctionnelle, causée par des organismes encapsulés tels que Streptococcus pneumoniae et Haemophilus influenzae. Tous les jours la prophylaxie de la pénicilline est le traitement le plus couramment utilisé pendant l'enfance, avec des hématologues traitement poursuivies indéfiniment. Les patients bénéficient aujourd'hui de la vaccination de routine pour H. influenzae, S. pneumoniae et Neisseria meningitidis.
- Stroke , qui peut résulter d'un rétrécissement progressif des vaisseaux sanguins, ce qui empêche l'oxygène d'atteindre la cerveau. Infarctus cérébral se produit chez les enfants et chez les adultes hémorragie cérébrale.
- AVC silencieux est un coup qui ne provoque pas de symptômes immédiats, mais est associée à des lésions au cerveau. AVC silencieux est probablement cinq fois plus communs que l'AVC symptomatique. Environ 10 à 15% des enfants atteints de drépanocytose souffrent coups, avec des AVC silencieux prédominant chez les patients plus jeunes.
- Lithiase biliaire (calculs biliaires) et cholécystite, qui peut résulter de excessive la production et la précipitation due à la bilirubine prolongée hémolyse.
- La nécrose avasculaire ( os nécrose aseptique) de la hanche et d'autres grandes articulations, ce qui peut se produire à la suite d'une ischémie.
- Diminution des réactions immunitaires en raison de hyposplénie (dysfonctionnement de la rate).
- Priapisme et infarctus du pénis.
- Ostéomyélite (infection osseuse bactérienne); la cause la plus commune de l'ostéomyélite dans la drépanocytose est Salmonella (surtout les sérotypes non-typique Salmonella typhimurium, Salmonella enteritidis, Salmonella choleraesuis et Salmonella paratyphi B), suivi par Staphylococcus aureus et les bacilles à Gram négatif entérique peut-être parce falciformation intravasculaire de l'intestin conduit à un infarctus ischémique inégale.
- La tolérance aux opioïdes, ce qui peut se produire comme une réponse physiologique normale à l'utilisation thérapeutique des opiacés. Dépendance aux opiacés se produit pas plus souvent chez les personnes atteintes de la drépanocytose que chez les autres personnes traitées avec les opiacés pour d'autres raisons.
- Nécrose papillaire aiguë dans les reins.
- Les ulcères de jambe.
- Dans les yeux, rétinopathie, rétinopathie proliférante, hémorragies vitreuses et décollement de la rétine, entraînant la cécité. Contrôles oculaires annuelles régulières sont recommandées.
- Pendant la grossesse, retard de croissance intra-utérin, spontanée l'avortement, et la pré-éclampsie.
- La douleur chronique: Même en l'absence de douleur aiguë vaso-occlusive, de nombreux patients ont une douleur chronique qui ne est pas signalé.
- L'hypertension artérielle pulmonaire (pression accrue sur le artère pulmonaire), conduisant à la souche sur le ventricule droit et un risque de insuffisance cardiaque; Les symptômes typiques sont de l'essoufflement, diminution de la tolérance et des épisodes de l'exercice syncope.
- Chronique insuffisance rénale due à La drépanocytose se manifeste-néphropathie avec l'hypertension (pression artérielle élevée), protéinurie (perte de protéines dans l'urine), hématurie (perte de globules rouges dans l'urine) et aggrave l'anémie. Si elle progresse de mettre fin à l'insuffisance rénale, il porte un mauvais pronostic.
Hétérozygotes
La forme hétérozygote ( trait drépanocytaire) est presque toujours asymptomatique, et la seule manifestation significative d'habitude est le défaut de concentration rénale présentant isosthenuria.
Physiopathologie
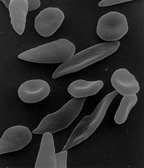
La drépanocytose est causée par un mutation ponctuelle dans la chaîne β-globine de l'hémoglobine, ce qui provoque l'acide aminé hydrophile de l'acide glutamique est remplacé par l'acide aminé hydrophobe valine à la sixième position. Le gène de la β-globine se trouve sur chromosome 11. L'association de deux sous-unités de type sauvage α-globine avec deux sous-unités de β-globine de l'hémoglobine S formes mutantes (HbS). Dans des conditions pauvres en oxygène (soit, à haute altitude, par exemple), l'absence d'un acide aminé polaire en position six de la chaîne β globine favorise la polymérisation non covalente (agrégation) de l'hémoglobine, ce qui fausse les globules rouges dans un forme de faucille et diminue leur élasticité.
La perte d'élasticité de la cellule rouge du sang est au centre de la physiopathologie de la maladie à hématies falciformes. Des globules rouges normaux sont assez élastique, ce qui permet aux cellules de se déformer pour passer à travers les capillaires. Dans la drépanocytose, la tension faible teneur en oxygène favorise rouge falciformation des globules et des épisodes répétés de falciformation endommagent la membrane cellulaire et diminuent l'élasticité de la cellule. Ces cellules ne parviennent pas à revenir à la forme normale quand la tension d'oxygène normale est rétablie. En conséquence, ces cellules sanguines rigides sont incapables de se déformer lors de leur passage à travers des capillaires étroits, conduisant à l'occlusion du vaisseau et ischémie.
L'anémie réelle de la maladie est causée par hémolyse, la destruction des globules rouges, en raison de leur misshape. Bien que le moelle osseuse tente de compenser en créant de nouveaux globules rouges, il ne correspond pas au taux de destruction. Globules rouges sains vivent généralement de 90 à 120 jours, mais les cellules de faucille ne survivent 10-20 jours.
Normalement, les humains ont hémoglobine A, qui se compose de deux alpha et deux chaînes bêta, l'hémoglobine A2, qui est constitué de deux chaînes alpha et delta et deux hémoglobine F, constitué de deux alpha et deux chaînes gamma dans leur corps. Parmi ceux-ci, l'hémoglobine A représente environ 96 à 97% de l'hémoglobine normale chez l'homme.
Génétique
Mutation du gène drépanocytaire est probablement apparue spontanément dans différentes zones géographiques, comme suggéré par l'analyse d'endonucléase de restriction. Ces variantes sont connus comme le Cameroun, le Sénégal, le Bénin, bantoue et l'Arabie-asiatique. Leurs sources d'importance clinique du fait que certains d'entre eux sont associés à des niveaux plus élevés HbF, par exemple, le Sénégal et variantes Arabie-asiatiques, et ont tendance à avoir une maladie plus bénigne.
Chez les personnes hétérozygote pour HGBS ( porteurs de falciformation l'hémoglobine), les problèmes de polymérisation sont mineures, car la normale allèle est capable de produire plus de 50% de l'hémoglobine. Chez les personnes homozygote pour HGBS, la présence de polymères à longue chaîne de HbS déformer la forme du globule rouge d'une lisse forme de beignet comme Ragged et plein de pointes, ce qui rend fragiles et sensibles à la rupture au sein capillaires. Les transporteurs ont des symptômes que si elles sont privées d'oxygène (par exemple, lors de l'ascension d'une montagne) ou pendant sévèrement déshydraté. La drépanocytose se produit lorsque le septième acide aminé (si la méthionine initiale est prise en compte), l'acide glutamique, est remplacé par la valine pour modifier sa structure et sa fonction. Valine est hydrophobe, ce qui provoque l'hémoglobine se effondrer sur lui-même à l'occasion. La structure ne est pas modifiée autrement. Lorsque suffisamment d'hémoglobine se effondre sur elle-même les globules rouges deviennent en forme de faucille.


Le défaut de gène est un connue mutation d'un seul nucléotides (voir Polymorphisme nucléotidique - SNP) (A à T) du gène de la β-globine, ce qui donne l'acide glutamique est substitué par valine à la position 6. L'hémoglobine S avec cette mutation est appelée HbS, par opposition à l'adulte normal HbA. La maladie génétique est due à la mutation d'un seul nucléotide, à partir d'un CTC CAC codon sur le brin matrice, qui est transcrite en un codon GUG. Ce est normalement une mutation bénigne, ne causant aucun effet apparent sur la secondaire, tertiaire ou structure quaternaire de l'hémoglobine normale dans des conditions de l'oxygène concentration. Qu'est-ce qu'il ne permet pour, dans des conditions de faible oxygène concentration, est le polymérisation de l'HbS lui-même. La forme désoxy de l'hémoglobine expose un patch hydrophobe sur la protéine entre les hélices E et F. Les résidus hydrophobes de la valine à la position 6 de la chaîne bêta de l'hémoglobine sont capables de se associer avec le patch hydrophobe, ce qui provoque l'hémoglobine S molécules à se agréger et former des précipités fibreux.
Le allèle responsable de la drépanocytose est autosomique récessive et peut être trouvé sur le bras court du chromosome 11. Une personne qui reçoit le gène défectueux de père et de mère développe la maladie; une personne qui reçoit une défectueux et un allèle sain reste en bonne santé, mais peut transmettre la maladie et est connu comme un transporteur. Si deux parents porteurs ont un enfant, il ya une chance de 1 sur 4 de leur enfant de développer la maladie et un 1 chance sur 2 de leur enfant étant juste un transporteur. Depuis la gène est incomplètement récessive, les transporteurs peuvent produire quelques globules rouges falciformes, pas assez pour causer des symptômes, mais assez pour donner la résistance au paludisme. Cela ne confère pas une immunité totale au paludisme; hétérozygotes sont encore capables de contracter le paludisme, mais leurs symptômes sont généralement moins graves. Pour cette raison, les hétérozygotes ont une ultérieure remise en forme de l'une des homozygotes. Ceci est connu comme avantage hétérozygote.
En raison de l'avantage adaptatif de l'hétérozygote, la maladie est encore répandue, surtout chez les personnes d'ascendance récente dans les zones frappées par le paludisme, comme l'Afrique , l' méditerranéenne , l'Inde et le Moyen-Orient . Le paludisme était endémique historiquement sud de l'Europe, mais il a été déclarée éradiquée dans le milieu du 20e siècle, à l'exception de rares cas sporadiques.
Le parasite du paludisme a un cycle de vie complexe et passe une partie de celui-ci dans les globules rouges. Dans un véhicule, la présence du parasite du paludisme entraîne les cellules rouges du sang avec de l'hémoglobine pour se rompre prématurément défectueux, ce qui rend le plasmodium incapables de se reproduire. En outre, la polymérisation de Hb affecte la capacité du parasite à digérer Hb en premier lieu. Par conséquent, dans les zones où le paludisme est un problème, les chances de survie des personnes augmentent effectivement si elles portent trait drépanocytaire (sélection pour l'hétérozygote).
Dans le États-Unis , où il n'y a pas de paludisme endémique, la prévalence de la drépanocytose chez les Noirs est plus faible (environ 0,25%) que dans l'Afrique de l'Ouest (environ 4,0%) et est en baisse. Sans le paludisme est endémique, la mutation drépanocytaire est purement désavantageuse et aura tendance à être sélectionné sur la population touchée par la sélection naturelle . Cependant, la communauté afro-américaine de la États-Unis est connu pour être le résultat d'un mélange important entre plusieurs groupes ethniques africains et non-africains, et représente également les descendants de survivants de l'esclavage et la traite négrière. Ainsi, un faible degré d'endogamie et, en particulier, la pression anormalement élevée de la santé-sélective au travers de l'esclavage peut être les explications les plus plausibles pour le plus faible prévalence de la drépanocytose (et, éventuellement, d'autres maladies génétiques) chez les Afro-Américains par rapport à Sous -Saharan Africains. Un autre facteur limitant la propagation des gènes drépanocytaires en Amérique du Nord est l'absence d'inclinations culturelles à la polygamie, qui permet aux hommes atteints de continuer à rechercher les enfants non atteints avec de multiples partenaires.

Héritage
Conditions drépanocytaires sont héritées des parents, de la même façon que le type de sang, la couleur des cheveux et la texture, la couleur des yeux, et d'autres traits physiques. Les types d'hémoglobine une personne fait dans les globules rouges dépendent de ce que les gènes de l'hémoglobine sont hérité de ses parents. Si un parent a la drépanocytose (SS) et l'autre a trait drépanocytaire alors il ya une chance de 50% d'avoir la maladie drépanocytaire d'un enfant et une chance d'avoir le trait drépanocytaire d'un enfant de 50%. Lorsque les deux parents ont trait drépanocytaire un enfant a une chance de 25% de la drépanocytose, comme le montre le diagramme.
Diagnostic
Dans du HBSS, le numération formule sanguine révèle le taux d'hémoglobine dans la plage de 6-8 g / dl avec une haute nombre de réticulocytes (comme la moelle osseuse pour compenser la destruction des cellules falciformes par la production de globules rouges). Dans d'autres formes de drépanocytose, les taux d'Hb ont tendance à être plus élevé. Un frottis sanguin peut montrer des caractéristiques de hyposplénie ( les cellules cibles et Howell-Jolly organes).
Falciformation des globules rouges, sur un film de sang, peut être induite par l'addition de le métabisulfite de sodium. La présence de l'hémoglobine faucille peut également être démontré avec le "test de solubilité faucille". Un mélange de l'hémoglobine S (Hb S) dans une solution réductrice (telle que dithionite de sodium) donne un aspect trouble, alors que Hb normale donne une solution claire.
Anormal formes d'hémoglobine peuvent être détectées sur électrophorèse de l'hémoglobine, une forme de électrophorèse sur gel sur lequel les différents types d'hémoglobine se déplacent à des vitesses variables. Hémoglobine drépanocytaire (HGBS) et hémoglobine C avec falciformation (HgbSC) -les deux plus courantes formes-peut être identifié à partir de là. Le diagnostic peut être confirmé par Chromatographie en phase liquide à haute performance (HPLC). Les tests génétiques est rarement pratiquée, comme d'autres enquêtes sont hautement spécifiques de HbS et Hbc.
Une crise drépanocytaire aiguë est souvent précipitée par l'infection. Par conséquent, une analyse d'urine pour détecter une infection occulte des voies urinaires, et la poitrine X-ray à regarder pour une pneumonie occulte doivent être effectuées régulièrement.
Les gens qui sont connus porteurs de la maladie subissent souvent conseil génétique avant d'avoir un enfant. Un test pour voir si l'enfant à naître a la maladie prend soit un sang échantillon de la foetus ou un échantillon de liquide amniotique. Depuis qu'il a pris un échantillon de sang d'un fœtus a de plus grands risques, ce dernier test est généralement utilisé.
Après la mutation responsable de cette maladie a été découvert en 1979, le US Air Force nécessaire candidats noirs de tester pour la mutation. Elle a rejeté 143 candidats parce qu'ils étaient porteurs, même si aucun d'eux ne avait la condition. Il a finalement retiré l'exigence, mais seulement après un stagiaire a intenté un procès.
Gestion
L'acide folique et de la pénicilline
Les enfants nés avec la drépanocytose subiront une surveillance étroite par le pédiatre et nécessiteront une gestion par un hématologue pour se assurer qu'ils restent en bonne santé. Ces patients auront une dose de 1 mg de l'acide folique par jour pour la vie. De la naissance à cinq ans, ils auront aussi à prendre la pénicilline par jour en raison du système immunitaire immature qui les rend plus sujettes aux maladies de la petite enfance.
chimioprophylaxie du paludisme
L'effet protecteur de trait drépanocytaire ne se applique pas aux personnes atteintes de la drépanocytose; en fait, ils sont particulièrement vulnérables au paludisme, puisque la cause la plus fréquente de crises douloureuses dans les pays paludisme est une infection par le paludisme. Il a donc été recommandé que les personnes atteintes de la drépanocytose vivant dans les pays paludisme devraient recevoir une chimioprophylaxie antipaludique pour la vie.
Crise vaso-occlusive
La plupart des personnes atteintes de drépanocytose ont épisodes intensément douloureux appelé crises vaso-occlusives. La fréquence, la gravité et la durée de ces crises, cependant, varient énormément. Crises douloureuses sont un traitement symptomatique avec des analgésiques ; gestion de la douleur nécessite l'administration d'opiacés à intervalles réguliers jusqu'à ce que la crise se est installée. Pour crises plus doux, un sous-groupe de patients à gérer sur AINS (tels que diclofénac ou naproxen). Pour des crises plus graves, la plupart des patients ont besoin de prise en charge hospitalière des opiacés par voie intraveineuse; analgésie dispositifs (PCA) de contrôlée par le patient sont couramment utilisés dans ce cadre. Diphenhydramine est également un agent efficace qui est souvent prescrit par les médecins pour aider à contrôler toute les démangeaisons associées à l'utilisation d'opioïdes.
Crise de poitrine aiguë
La direction est semblable à une crise vaso-occlusif, avec l'ajout d'antibiotiques (habituellement un macrolide ou quinolone, depuis la paroi cellulaire déficiente ["atypiques"] bactéries sont considérés comme contribuant au syndrome), la supplémentation en oxygène pour hypoxie, et l'observation étroite. Si pulmonaire infiltrer aggravent ou l'augmentation des exigences de l'oxygène, simple transfusion sanguine ou exsanguinotransfusion est indiquée. Celle-ci implique le remplacement d'une partie importante de la masse des globules rouges patients pour les globules rouges normales, ce qui diminue le pour cent de l'hémoglobine S dans le sang du patient.
Hydroxyurée
Le premier médicament approuvé pour le traitement causal de la drépanocytose, hydroxyurée, a été montré pour diminuer le nombre et la gravité des attaques dans une étude en 1995 (Charache et al.) et montré à éventuellement augmenter le temps de survie dans une étude en 2003 (Steinberg et al.). Ceci est réalisé, en partie, en réactivant la production d'hémoglobine fœtale en place de l'hémoglobine S qui provoque la drépanocytose. L'hydroxyurée a déjà été utilisé en tant que agent de chimiothérapie, et il est à craindre que l'utilisation à long terme peut être nocive, mais ce risque a été montré pour être soit absent ou très faible et il est probable que les avantages l'emportent sur les risques.
La thérapie transfusionnelle
Les transfusions sanguines sont souvent utilisés dans le traitement de la maladie à hématies falciformes dans les cas aigus et à prévenir les complications en diminuant le nombre de globules rouges (RBC) qui peut faucille en ajoutant des globules rouges normaux. Chez les enfants de globules rouges chronique prophylactique (RBC) thérapie transfusionnelle a été montré pour être efficace dans une certaine mesure pour réduire le risque d'un premier AVC ou accident vasculaire cérébral silencieux quand Doppler transcrânien (DTC) échographie montre une augmentation des anomalies cérébrales vitesses d'écoulement du sang. Dans ceux qui ont subi un événement de course avant elle réduit également le risque de récidive d'AVC et AVC silencieux supplémentaires.
Greffes de moelle osseuse
Greffes de moelle osseuse ont prouvé leur efficacité chez les enfants. Greffes de moelle osseuse sont le seul remède connu pour SCD.
Pronostic
Environ 90% des patients survivent à 20 ans, et près de 50% survivent au-delà de la cinquième decade.In 2001, selon une étude, les quelque survie moyenne pour les patients drépanocytaires était de 53 ans pour les hommes et 58 ans pour les femmes SCD homozygote.
Épidémiologie
La fréquence la plus élevée de la drépanocytose se trouve dans les régions tropicales, en particulier en Afrique subsaharienne, en Inde et au Moyen-Orient. Migration d'importantes populations de ces zones à forte prévalence de pays à faible prévalence en Europe a considérablement augmenté au cours des dernières décennies et, dans certains pays européens la drépanocytose a maintenant dépassé conditions génétiques plus familiers tels que l'hémophilie et la mucoviscidose .
Afrique
Trois quarts des cas drépanocytaires se produisent en Afrique. Une récente OMS rapport estime qu'environ 2% des nouveau-nés au Nigeria ont été touchés par la drépanocytose, donnant un total de 150 000 enfants touchés nés chaque année dans le seul Nigéria. La fréquence porteuse comprise entre 10% et 40% en Afrique équatoriale, diminuant à 1-2% sur la côte nord-africaine et <1% en Afrique du Sud.
États Unis
Le la prévalence de la maladie dans le États-Unis est d'environ 1 sur 5000, qui touche principalement les Américains d'origine africaine subsaharienne, selon le National Institutes of Health. Aux États-Unis, environ 1 sur 500 enfants afro-américains et 1: 36 000 enfants hispano-américains nés auront la drépanocytose. On estime que la drépanocytose (SCD) affecte 90 000 Américains. La plupart des nourrissons atteints de drépanocytose nés aux États-Unis sont maintenant identifiés par le dépistage néonatal de routine. Quarante-quatre États ainsi que le District de Columbia, Puerto Rico et les îles Vierges fournissent actuellement dépistage néonatal universel pour SCD. Trait drépanocytaire se produit entre environ 01h12 Afro-Américains et une: 100 Hispano-Américains. On estime que 2,5 millions d'Américains sont porteurs hétérozygotes pour le trait drépanocytaire.
France
Dans l'Europe , une forte prévalence de la maladie a été observée chez France . En raison de la croissance de la population dans les régions d'Afrique-Caraïbes de France d'outre-mer, et maintenant essentiellement de l'immigration du Nord et Afrique sub-saharienne en France métropolitaine, la drépanocytose est devenue un problème majeur de santé publique en France. SCD est devenu la maladie génétique la plus répandue dans ce pays, avec une prévalence à la naissance globale de 1/2415 en France métropolitaine, à l'avance phénylcétonurie (1/10862), congénitale hypothyroïdie (1/3132), congénitale hyperplasie surrénale (1 / 19,008) et la mucoviscidose (1/5014) pour la même période de référence. En 2010, 31,5% de tous les nouveau-nés en France métropolitaine (253 466) 805 958 sur avaient des parents proviennent d'une région définie «à risque» (principalement l'Afrique et les départements et territoires d'outre-mer de la France) et ont été criblés de la drépanocytose. 341 nouveau-nés atteints de drépanocytose et 8744 ont été trouvés porteurs hétérozygotes représentant 1,1% de tous les nouveau-nés en France métropolitaine. Le quartier de Paris métropolitaine ( Île-de-France) est la région qui compte le plus grand nombre de personnes à risque vraisemblablement plus élevé de SCD. En effet, 60% de tous les nouveau-nés dans ce domaine en 2010 avaient des parents sont originaires d'une région définie comme «à risque» et ont été criblées pour SCD. Le deuxième plus grand nombre de risques au-est Provence-Alpes-Côte d'Azur à près de 43,2% et le nombre le plus bas est en Bretagne à 5,5%. Ce pourcentage était de 19% en 2000 et a augmenté de 65%.
Royaume-Uni
Au Royaume-Uni, 1 bébé dans chaque 2000 est né avec cette condition.
Moyen-Orient
En Arabie Saoudite environ 4,2% de la population porter le trait drépanocytaire et 0,26% ont la drépanocytose. La prévalence la plus élevée est dans la province orientale, où environ 17% de la population porteuse du gène et 1,2% ont la drépanocytose. En 2005 en Arabie Saoudite un test prénuptial obligatoire y compris électrophorèse HB a été lancé et vise à réduire l'incidence de la SCD et thalassémie.
Inde
La drépanocytose est répandue dans de nombreuses régions de l'Inde, où la prévalence a varié de 9,4 à 22,2% dans les zones endémiques.
Histoire
Cette collection de résultats cliniques était inconnu jusqu'à l'explication des cellules falciformes en 1910 par le cardiologue Chicago et professeur de médecine James Herrick (1861-1954), dont le stagiaire Ernest Edward Irons (1877-1959) trouvé cellules "particulières allongées et en forme de faucille" dans le sang de Walter Clement Noel, 20 ans, étudiant en médecine dentaire de première année de la Grenade, après Noel a été admis à l'hôpital presbytérien de Chicago décembre 1904 souffrant de l'anémie .
Noel a été réadmis à plusieurs reprises au cours des trois prochaines années pour "rhumatisme musculaire" et "attaques" bilieux. Noel a terminé ses études et est retourné à la capitale de la Grenade (Saint-Georges) à pratiquer dentisterie. Il est mort d' une pneumonie en 1916 et est enterré dans le cimetière catholique Sauteurs dans le nord de la Grenade. Compte rendu publié de Herrick inclus illustrations, mais le premier slide disponibles montrant cellules falciformes est celle d'une autopsie d'un soldat avec trait faucille 1918, d'abord en revue seulement 92 ans plus tard.
La maladie a été nommé «drépanocytose» par Verne Mason en 1922, puis un médecin résident au Hôpital Johns Hopkins. Toutefois, certains éléments de la maladie ont été reconnus plus tôt: Un article dans le Journal sud de pharmacologie médicale en 1846 décrit l'absence de la rate dans le autopsie d'un esclave en fuite. La littérature médicale africaine a signalé cette condition dans les années 1870, quand il était connu localement comme ogbanjes ("enfants qui vont et viennent») en raison du taux de mortalité infantile très élevé causé par cette condition. Une histoire de la condition suivi des rapports de 1670 dans une famille ghanéenne.
Linus Pauling et ses collègues ont été les premiers, en 1949, pour démontrer que la drépanocytose se produit en raison d'une anomalie dans la molécule d'hémoglobine. Ce était la première fois qu'une maladie génétique est liée à une mutation d'une protéine spécifique, un jalon dans le l'histoire de la biologie moléculaire, et il a été publié dans leur article " Sickle Cell anémie, une maladie moléculaire ".